

| 產(chǎn)品編號 | bs-4105R |
| 英文名稱 | Filamin A Rabbit pAb |
| 中文名稱 | 細(xì)絲蛋白A抗體 |
| 別 名 | ABP 280; ABP 280 like protein; ABP-280; ABP280A; ABPA; Actin binding like protein; Actin binding protein 280; Actin-binding protein 280; alpha filamin; alpha-filamin; APBX; cb967; Dilp2; Endothelial actin binding protein; Endothelial actin-binding protein; Filamin 1; Filamin A alpha actin binding protein 280; Filamin A; Filamin-1; Filamin-A; FLN; FLN-A; FLN1; FLNA; FLNA_HUMAN; FMD; MNS; NHBP; Non muscle filamin; Non-muscle filamin; OPD; OPD1; OPD2. |
| 研究領(lǐng)域 | 心血管 免疫學(xué) 神經(jīng)生物學(xué) 結(jié)合蛋白 內(nèi)皮細(xì)胞 |
| 抗體來源 | Rabbit |
| 克隆類型 | Polyclonal |
| 克 隆 號 | |
| 交叉反應(yīng) | Human (predicted: Mouse,Rat,Rabbit,Pig,Cow,Dog,Horse) |
| 產(chǎn)品應(yīng)用 | WB=1:500-2000
not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 理論分子量 | 291 kDa |
| 檢測分子量 | |
| 細(xì)胞定位 | 細(xì)胞漿 |
| 性 狀 | Liquid |
| 濃 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human Filamin A: 1851-1960/2647 |
| 亞 型 | IgG |
| 純化方法 | affinity purified by Protein A |
| 緩 沖 液 | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| 保存條件 | Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles. |
| 注意事項 | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| PubMed | PubMed |
| 產(chǎn)品介紹 |
The protein encoded by this gene is an actin-binding protein that crosslinks actin filaments and links actin filaments to membrane glycoproteins. The encoded protein is involved in remodeling the cytoskeleton to effect changes in cell shape and migration. This protein interacts with integrins, transmembrane receptor complexes, and second messengers. Defects in this gene are a cause of several syndromes, including periventricular nodular heterotopias (PVNH1, PVNH4), otopalatodigital syndromes (OPD1, OPD2), frontometaphyseal dysplasia (FMD), Melnick-Needles syndrome (MNS), and X-linked congenital idiopathic intestinal pseudoobstruction (CIIPX). Two transcript variants encoding different isoforms have been found for this gene.[provided by RefSeq, Mar 2009] Function: Promotes orthogonal branching of actin filaments and links actin filaments to membrane glycoproteins. Anchors various transmembrane proteins to the actin cytoskeleton and serves as a scaffold for a wide range of cytoplasmic signaling proteins. Interaction with FLNA may allow neuroblast migration from the ventricular zone into the cortical plate. Tethers cell surface-localized furin, modulates its rate of internalization and directs its intracellular trafficking. Subunit: Interacts with PDLIM2 (By similarity). Homodimer. Interacts with FCGR1A, FLNB, FURIN, HSPB7, INPPL1, KCND2, MYOT, MYOZ1, ARHGAP24, PSEN1, PSEN2 and ECSCR. Interacts also with various other binding partners in addition to filamentous actin. Interacts (via N-terminus) with MIS18BP1 (via N-terminus). Interacts (via N-terminus) with TAF1B. Interacts with TMEM67 (via C-terminus) and MKS1. Subcellular Location: Cytoplasm, cell cortex. Cytoplasm, cytoskeleton. Tissue Specificity: Ubiquitous. Post-translational modifications: Phosphorylated upon DNA damage, probably by ATM or ATR. Phosphorylation extent changes in response to cell activation. DISEASE: Defects in FLNA are the cause of periventricular nodular heterotopia type 1 (PVNH1) [MIM:300049]; also called nodular heterotopia, bilateral periventricular (NHBP or BPNH). PVNH is a developmental disorder characterized by the presence of periventricular nodules of cerebral gray matter, resulting from a failure of neurons to migrate normally from the lateral ventricular proliferative zone, where they are formed, to the cerebral cortex. PVNH1 is an X-linked dominant form. Heterozygous females have normal intelligence but suffer from seizures and various manifestations outside the central nervous system, especially related to the vascular system. Hemizygous affected males die in the prenatal or perinatal period. Defects in FLNA are the cause of periventricular nodular heterotopia type 4 (PVNH4) [MIM:300537]; also known as periventricular heterotopia Ehlers-Danlos variant. PVNH4 is characterized by nodular brain heterotopia, joint hypermobility and development of aortic dilation in early adulthood. Defects in FLNA are the cause of otopalatodigital syndrome type 1 (OPD1) [MIM:311300]. OPD1 is an X-linked dominant multiple congenital anomalies disease mainly characterized by a generalized skeletal dysplasia, mild mental retardation, hearing loss, cleft palate, and typical facial anomalies. OPD1 belongs to a group of X-linked skeletal dysplasias known as oto-palato-digital syndrome spectrum disorders that also include OPD2, Melnick-Needles syndrome (MNS), and frontometaphyseal dysplasia (FMD). Remodeling of the cytoskeleton is central to the modulation of cell shape and migration. FLNA is a widely expressed protein that regulates re-organization of the actin cytoskeleton by interacting with integrins, transmembrane receptor complexes and second messengers. Males with OPD1 have cleft palate, malformations of the ossicles causing deafness and milder bone and limb defects than those associated with OPD2. Obligate female carriers of mutations causing both OPD1 and OPD2 have variable (often milder) expression of a similar phenotypic spectrum. Defects in FLNA are the cause of otopalatodigital syndrome type 2 (OPD2) [MIM:304120]; also known as cranioorodigital syndrome. OPD2 is a congenital bone disorder that is characterized by abnormally modeled, bowed bones, small or absent first digits and, more variably, cleft palate, posterior fossa brain anomalies, omphalocele and cardiac defects. Defects in FLNA are the cause of frontometaphyseal dysplasia (FMD) [MIM:305620]. FMD is a congenital bone disease characterized by supraorbital hyperostosis, deafness and digital anomalies. Defects in FLNA are the cause of Melnick-Needles syndrome (MNS) [MIM:309350]. MNS is a severe congenital bone disorder characterized by typical facies (exophthalmos, full cheeks, micrognathia and malalignment of teeth), flaring of the metaphyses of long bones, s-like curvature of bones of legs, irregular constrictions in the ribs, and sclerosis of base of skull. Defects in FLNA are the cause of X-linked congenital idiopathic intestinal pseudoobstruction (CIIPX) [MIM:300048]. CIIPX is characterized by a severe abnormality of gastrointestinal motility due to primary qualitative defects of enteric ganglia and nerve fibers. Affected individuals manifest recurrent signs of intestinal obstruction in the absence of any mechanical lesion. Defects in FLNA are the cause of FG syndrome type 2 (FGS2) [MIM:300321]. FG syndrome (FGS) is an X-linked disorder characterized by mental retardation, relative macrocephaly, hypotonia and constipation. Defects in FLNA are the cause of terminal osseous dysplasia (TOD) [MIM:300244]. A rare X-linked dominant male-lethal disease characterized by skeletal dysplasia of the limbs, pigmentary defects of the skin and recurrent digital fibroma during infancy. A significant phenotypic variability is observed in affected females. Defects in FLNA are the cause of cardiac valvular dysplasia X-linked (CVDX) [MIM:314400]. A rare X-linked heart disease characterized by mitral and/or aortic valve regurgitation. The histologic features include fragmentation of collagenous bundles within the valve fibrosa and accumulation of proteoglycans, which produces excessive valve tissue leading to billowing of the valve leaflets. Note=Defects in FLNA may be a cause of macrothrombocytopenia, a disorder characterized by subnormal levels of blood platelets. Blood platelets are abonormally enlarged. Similarity: Belongs to the filamin family. Contains 1 actin-binding domain. Contains 2 CH (calponin-homology) domains. Contains 24 filamin repeats. SWISS: P21333 Gene ID: 2316 Database links: Entrez Gene: 2316 Human Entrez Gene: 192176 Mouse Omim: 300017 Human SwissProt: P21333 Human SwissProt: Q8BTM8 Mouse Unigene: 195464 Human Unigene: 295533 Mouse Unigene: 4213 Rat 內(nèi)皮細(xì)胞肌動蛋白結(jié)合蛋白(肌動結(jié)合蛋白樣蛋白) |
| 產(chǎn)品圖片 |
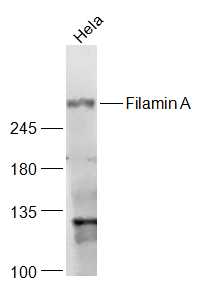
Sample:
Hela(Human) Cell Lysate at 30 ug
Primary: Anti-Filamin A (bs-4105R) at 1/1000 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 291 kD
Observed band size: 291 kD
|




